Sobre el síndrome
El Síndrome de Rokitansky, también conocido como Síndrome de Mayer-Rokitansky KusterHauser o Síndrome MRKH, lleva este nombre en honor a los médicos investigadores August Franz Josef Karl Mayer (1787–1865), Carl Freiherr von Rokitansky (1804–1878), Hermann Küster (1879–1964) y Georges Andre Hauser (1921–2009). Tal vez algún día podamos llamarlo simplemente Síndrome Roki.
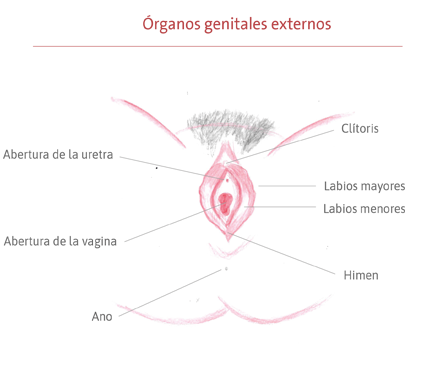
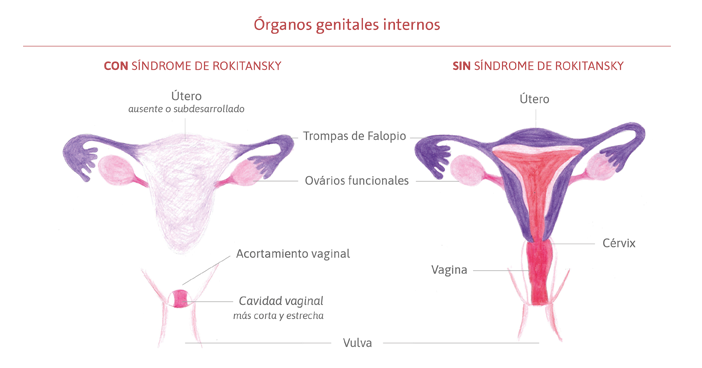
Es un síndrome congénito de causa aún desconocida que afecta el sistema reproductivo femenino durante su formación, aún en los primeros meses de vida fetal. Los órganos genitales comienzan a crecer, pero no se desarrollan completamente, afectando principalmente la formación del útero y el canal vaginal. En la mayoría de los casos, el útero está ausente o es muy pequeño y el canal vaginal (el camino entre el útero y la vulva) es más corto y más estrecho de lo habitual, pudiendo también estar ausente. La genitalia externa, la vulva (clítoris, uretra, labios menores y mayores, himen) y el ano tienen un desarrollo normal, al igual que los ovarios y las trompas de Falopio (conducto que lleva el óvulo del ovario al útero).
El síndrome se clasifica en dos tipos:
- Tipo I, con afectación aislada de los órganos reproductores, con una incidencia de una de cada 5 mil mujeres
- Tipo II, con asociaciones de alteraciones sistémicas en otros órganos, con una incidencia de una de cada 10 mil a 15 mil mujeres, siendo casos aún más raros.