About the syndrome
The Rokitansky Syndrome, also known as Mayer-Rokitansky-Küster-Hauser Syndrome or MRKH Syndrome, is named after the medical researchers August Franz Josef Karl Mayer (1787-1865), Carl Freiherr von Rokitansky (1804-1878), Hermann Küster (1879-1964), and Georges Andre Hauser (1921-2009). Perhaps one day we can simply call it Roki Syndrome.
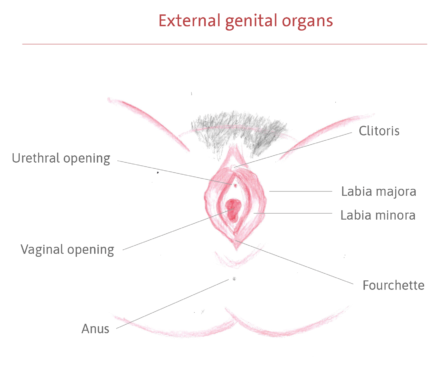
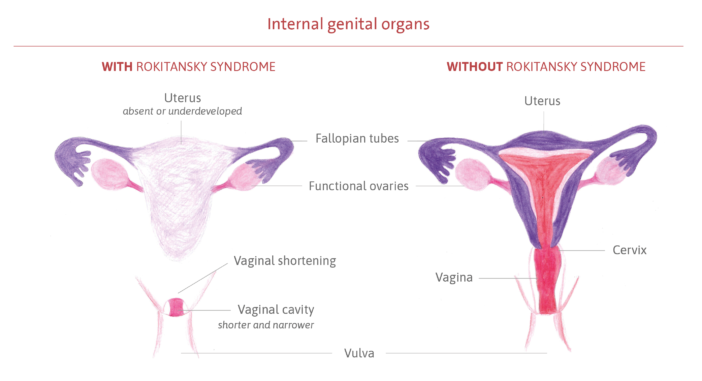
It is a congenital syndrome of unknown cause that affects the female reproductive system during its formation, in the first few months of fetal life. The genital organs begin to grow, but do not fully develop, mainly affecting the formation of the uterus and vaginal canal. In most cases, the uterus is absent or very small, and the vaginal canal (the pathway between the uterus and the vulva) is shorter and narrower than usual, and may also be absent. The external genitalia, the vulva (clitoris, urethral opening, labia minora and majora, hymen), and anus develop normally, as do the ovaries and fallopian tubes (the duct that carries the egg from the ovary to the uterus).
The syndrome is classified into two types:
- Type I, with isolated involvement of the reproductive organs, with an incidence of one in every 5,000 women;
- Type II, with associations of systemic alterations in other organs, with an incidence of one in every 10,000 to 15,000 women, with even rarer cases.